
Preparation and activity study for Organoruthenium electrocatalysts for water and ammonia oxidation – towards sustainable Hydrogen production
Electrochemical Oxidation:
It is more and more evident that humankind has to develop new cleaner energy sources to replace obsolete fossil fuels. Many see hydrogen as such fuel for the future. However, its volatility, sensitivity, high liquefaction and transportation costs hinder its wide-scale use. These obstacles could be solved by developing an electrochemical cell that liberates hydrogen from a possibly benign compound for direct on-site use. Below, two processes (Water and Ammonia oxidation, Figure 2)) are introduced to illustrate this concept. The significant advantage here is their complementarity, meaning that the mechanistic rationale and catalysts from one process are directly transferable to the other one
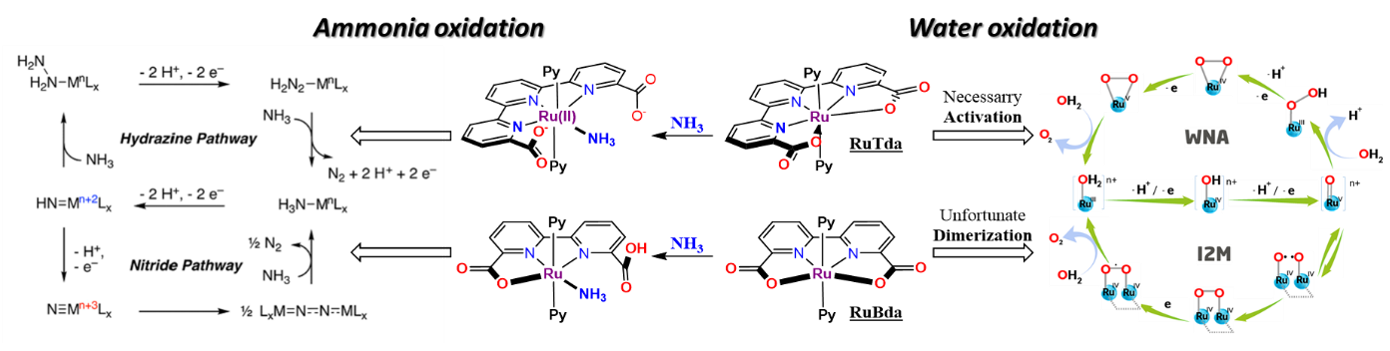
Figure 1: Comparison of catalytic mechanisms of RuTda (monomolecular) and RuBda (dimeric); Left: Ammonia oxidation; Right: Water oxidation
Water oxidation catalysis (WOC)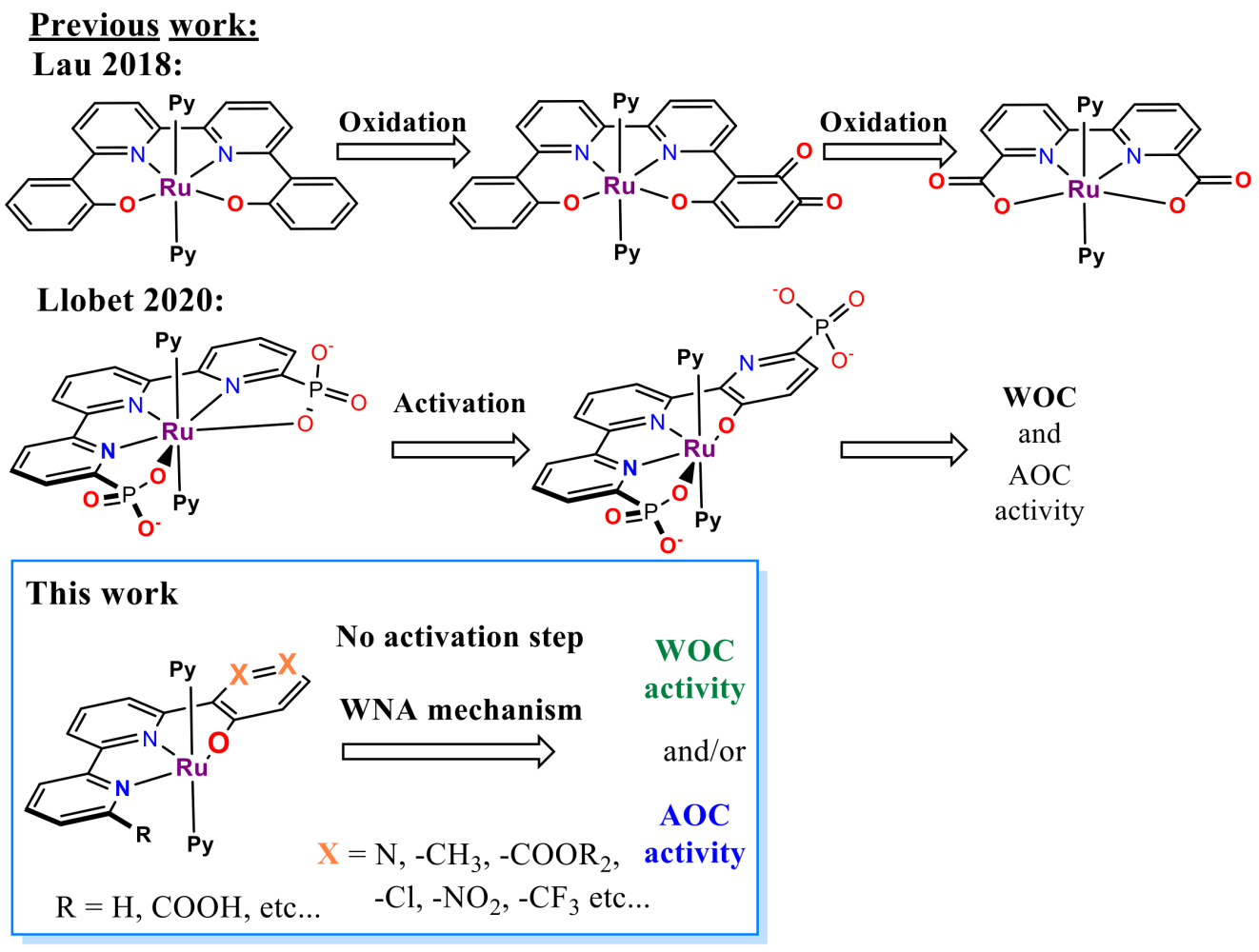
The most pursued concept relies naturally on H2O combined with a water oxidation catalyst. Water oxidation has been a holy grail of green renewable energy since the term was coined. The sheer abundance and complete harmlessness make water a dream source of energy. However, even after years of research, there is no economically viable commercial utilisation. In this regard, the closest to an application are ruthenium-based electrocatalysts with their low overpotentials, comfortable working window in a whole pH scale and relatively high stability.[1] In contrast, first-row transition metal-based WOC catalysts suffer from their reliance on extreme pH and high overpotentials while displaying low life duration (Low TOFs and TONs).[1] From an economic point of view, if ruthenium catalysts are stable in the long term, the initial high investment can be balanced with a sustained, steady and long-lasting return.
The field of ruthenium water oxidation catalysts has seen striking development in recent years with the rise of two prominent families - the RuBda[2] (Bda = 2,2’-Bipyridine-6,6’-dicarboxylic acid) and RuTda[3] (Tda = 2,2':6',2''-Terpyridine]-6,6''-dicarboxylic acid) (Figure 2).
The literature sources show already a wide range of studies dealing with derivatisation, surface anchoring and applications.[1] However, each of these families still suffers from its own critical weaknesses preventing their practical use. For example, RuTda, the most promising catalyst for application, as it works through monomolecular water nucleophilic attack (WNA) mechanism (Figure 2), needs a bulk electrolytic activation under strongly basic conditions to reach its peak performance.[3] In comparison, RuBda does not need any activation but works through a dimeric (Interaction of two M-O units – I2M) mechanism, which renders this catalyst unsuitable for surface anchoring and consequently unsuitable for practical applications. The surface anchoring increases the active concentration of the catalyst in the diffusion layer at the electrode and improves electron transfer and the overall robustness of the electrochemical set-up.
It was shown that the structural analogue of RuTda, the RutPa undergoes partial oxidation under the catalytic conditions giving a new pyridine-3-oxo species (Figure 3), which is assumed to be the actual active catalyst.[4] Building upon the structural similarities, introducing the hydroxyl group into the terpyridine offers good chances to eliminate the independent activation step.
Ammonia oxidation catalysis (AOC)
As an alternative to water, ammonia has risen in the last two years as a challenger for future hydrogen source. Its advantages are low thermodynamic constraints for electrocatalytic oxidation (NH3 -0.092 V vs H2O 1.23V vs NHE), the high energy density of liquid ammonia (1.5x of liquid H2) and existing worldwide processing infrastructure[5]. Low oxidative potential means lower operational costs and cheaper devices. Also, the existing infrastructure indicates that with any future progress in NH3 synthesis, this approach can quickly become technologically feasible. Coincidentally, it has been shown by several studies that the same complexes active in WOC can also be applied in AOC, meaning that discoveries from one process can be directly applied to the other and vice versa.
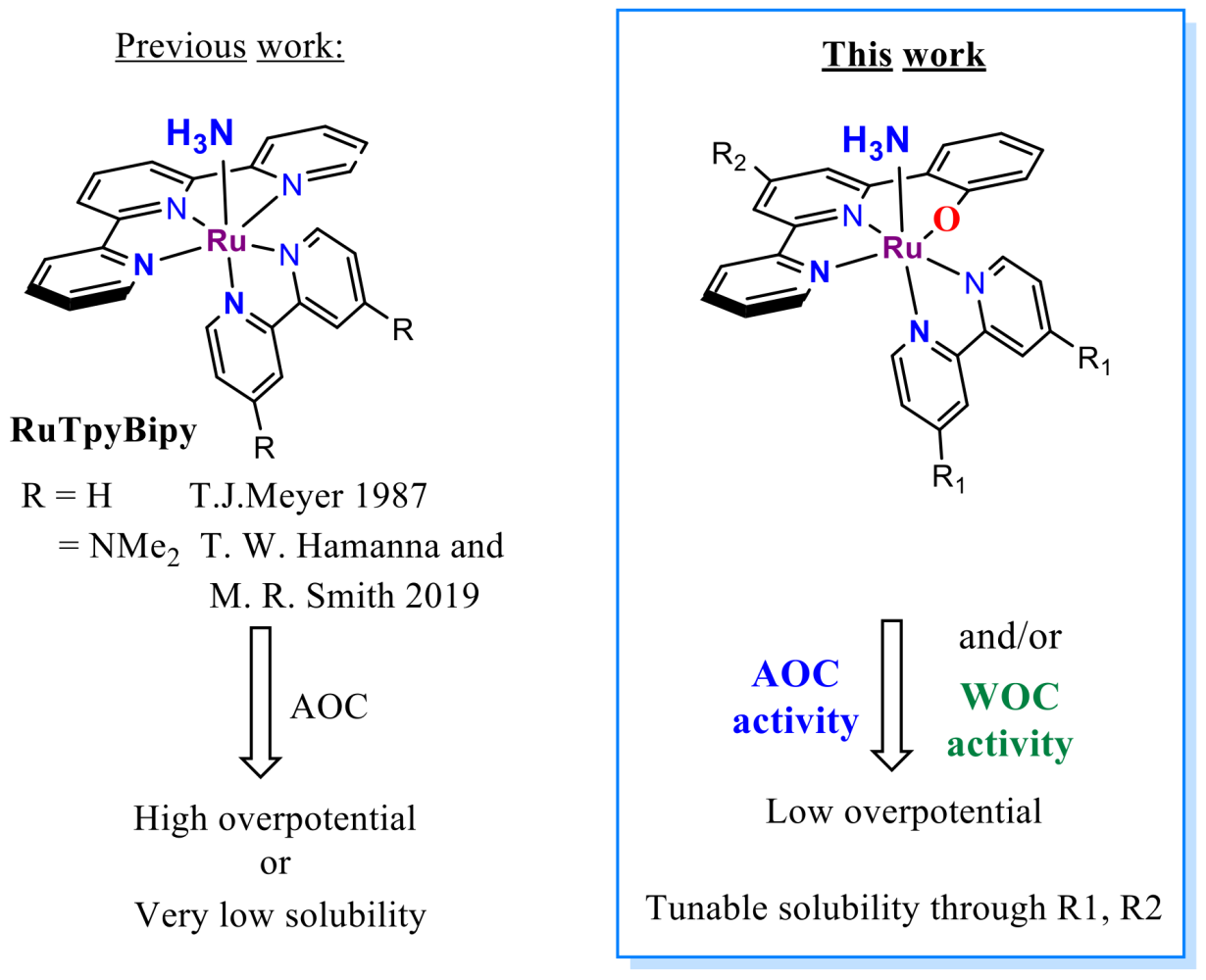
The most notable discoveries in this field, both from 2019, are Hamannna and Smith's improvement of the old RuTerpyBipy[6][7] motive (RuTerpy[Bipy(NMe2)2]) and Nishibayashi's RuBda study[8] (Figure 4). The RuBda again works through a disadvantageous bimolecular mechanism (Figure 2) relying on the dimerisation of Ru-bound nitride units, thus effectively thwarting further practical applications.
In comparison, Hamanna's and Smith's RuTerpy[Bpy(NMe2)2] (Figure 4) catalyst works through an advantageous monomolecular mechanism relying on the nucleophilic attack of ammonia to Ru-bound imine. However, the –NMe2 used to shift electrochemical potentials significantly reduced the solubility of the complex to the point where it prevented any further development. [6] The aim here is, again, through the introduction of the hydroxyl group keep the low overpotential[9] while maintaining the good solubility of the catalyst. Furthermore, it was shown that the exchange of N for –O- in Ru(Bpy)3 type complex leads to a favourable cathodic shift for more than 0.5 V at RuII/III transition. [9]
1) Ligands and the Complexes
At first, two distinguished families of ligands and complexes with coordinated aryl/pyridyl oxide will be synthesised and tested in WOC and AOR accordingly. (Figure 5)
The first catalyst family will be built over the RuTerpyBipy motive using the bipyridine-phenoxide (BPPO) (1, Figure 4 and 5b) ligand bearing no auxiliary substituents (R1 = -H, Figure 5b). Later the substitution on BPPO or bipyridine outer rim can fine-tune its physicochemical properties (R1, R2 Figure 4). Although the RuTerpyBpy design mainly targets AOC reactivity, it also bears considerable potential for WOC.[10]
In addition to hydroxyl, the second family will also have the positions 3, 4- (in regard to hydroxyl, Figures 3 and 2 in Figure 5b) occupied either with heteroatoms, decorated with functional groups (e.g. COOR, CF3 etc.) or a combination of both. The inherent properties of Ruthenium ensure that the hydroxyl coordinates to the metal centre achieving N2O2 coordination vicinity. The ability of pyridine to rotate[4] provides an opportunity for auto-corrective hydroxyl coordination in the case of the nitrogen coordinates first (Figure 3). Furthermore, the additional substituents will protect the aromatic core from further oxidation, thus preventing the decomposition described by Lau.[11] The resemblance to RuTda predetermines this type of complex for WOC; however, the preliminary results also give confidence for its complementary activity in AOC.
2) Electrochemical characterisation, mechanistic studies and catalytic performance assessment
After the synthesis, all the compounds will be subjected to electrochemical analysis (CV, DPV, CPE, etc.) and assessment. The elegance of the electrochemical methods is in their experimental simplicity while still being highly informative. By measuring CVs at different scan rates, concentrations or variations in other conditions, it is possible to get highly precise information about TON and TOF as well as other kinetic and mechanistic data. Later, through the bulk electrolysis, it will be possible to assess the stability of our catalyst under working conditions as well as to quantify the amount of produced gasses (O2 or N2 and H2) via analysis of reaction headspace with gas chromatography or other established analytic method (e.g. Clark electrode).
3) Enhancement and tuning of our catalysts
The described design of the catalysts allows simple modification of the ligands and the metal core for alternative catalytic processes. Despite many possibilities, the most thrilling one is the addition of the pyrene moiety. This will allow us to anchor our catalysts through π-π stacking on the graphitic surfaces and, consequently, onto the electrodes through the drop-casting deposition. (Figure 6). This simple adjustment can enhance the electron transfer from the electrode to the catalyst, increase its effective concentration in the active diffusion layer and lead to stable, reusable electrodes for long-term applications. [12]
Further options include introducing other solubilising and electron-donating/withdrawing groups to tune the physicochemical properties of the complexes. Lastly, the ligand invites for complexation with different metal cations such as Os, Ir or first-row transition metals such as Mn, Fe. This variability can open the door to new processes and serve as both a backup strategy and added valuation for the ligands.
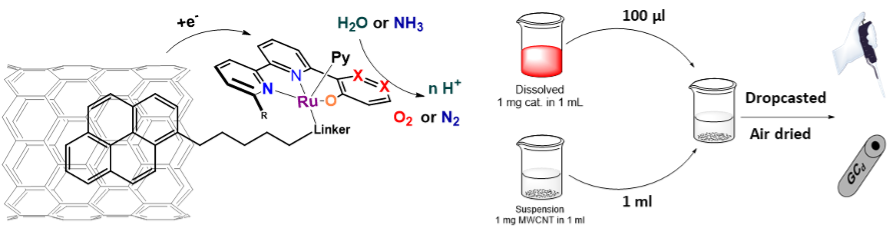
Figure 4: Illustration of the catalyst anchoring via π-π stacking and visualisation of the drop-casting process
[1] R. Matheu, P. Garrido-Barros, M. Gil-Sepulcre, M. Z. Ertem, X. Sala, C. Gimbert-Suriñach, A. Llobet, Nat. Rev. Chem. 2019, 3, 331–341.
[2] L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet, L. Sun, Nat. Chem. 2012, 4, 418–423.
[3] R. Matheu, M. Z. Ertem, J. Benet-Buchholz, E. Coronado, V. S. Batista, X. Sala, A. Llobet, J. Am. Chem. Soc. 2015, 137, 10786–10795.
[4] N. Vereshchuk, R. Matheu, J. Benet-Buchholz, M. Pipelier, J. Lebreton, D. Dubreuil, A. Tessier, C. Gimbert-Suriñach, M. Z. Ertem, A. Llobet, J. Am. Chem. Soc. 2020, 142, 5068–5077.
[5] N. M. Adli, H. Zhang, S. Mukherjee, G. Wu, J. Electrochem. Soc. 2018, 165, J3130–J3147.
[6] F. Habibzadeh, S. L. Miller, T. W. Hamann, M. R. Smith, Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 2849–2853.
[7] M. S. Thompson, T. J. Meyer, J. Am. Chem. Soc. 1981, 103, 5577–5579.
[8] K. Nakajima, H. Toda, K. Sakata, Y. Nishibayashi, Nat. Chem. 2019, 11, 702–709.
[9] B. M. Holligan, J. C. Jeffery, M. K. Norgett, E. Schatz, M. D. Ward, J. Chem. Soc. Dalt. Trans. 1992, 3345–3351.
[10] R. Matheu, M. Z. Ertem, C. Gimbert-Suriñach, J. Benet-Buchholz, X. Sala, A. Llobet, ACS Catal. 2017, 7, 6525–6532.
[11] Y. Liu, G. Chen, S. M. Yiu, C. Y. Wong, T. C. Lau, ChemCatChem 2018, 10, 501–504.
[12] J. Creus, R. Matheu, I. Peñafiel, D. Moonshiram, P. Blondeau, J. Benet-Buchholz, J. García-Antón, X. Sala, C. Godard, A. Llobet, Angew. Chemie - Int. Ed. 2016, 55, 15382–15386.