
Group profile
Computational methods of quantum mechanics and chemical thermodynamics represent powerful and widely used tools for prediction and interpretation of properties and behavior of chemical substances. The main goal of our research is the first-principle calculation of thermodynamic properties of inorganic materials as well as their application in phase and chemical equilibria modeling and phase diagrams construction.
Research focus
|
 |
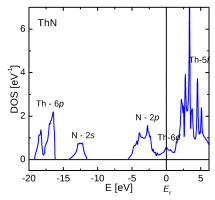 |
| Electron denstity and density of states of ThN | ||
Studied materials
|
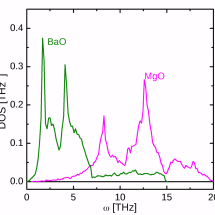
Phonon spectrum of MgO and BaO |
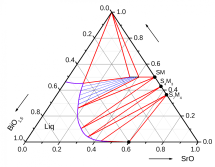
Phase diagram of BiSrMnO system |
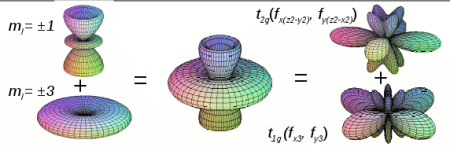
Analysis of valence states U-5f in UO2
Staff
David Sedmidubský
PhD students: J. Mokrý, J. Cajzl
Collaboration: J. Leitner (DME UCT), J. Macháček (Dep.of Ceramics UCT), K. Knížek (IP ASCR)
R.J.M. Konings, O.Beneš (JRC-EC Karlsruhe)
Equipment
|
Hardware: computational cluster 4x Intel Core i7 / 8 Gb RAM / 500 Gb HD, openSUSE 12.2 Software: WIEN2k – DFT program for electron structure calculation of crystalline solids using full potential LAPW / APW+lo technique VASP - DFT program for electron structure calculation of crystalline solids using pseudopotential technique Phonon – program for calculation of phonon spectra and related thermodynamic properties of solids |
 |
 |
 |
 |
Selected publications
- D.Sedmidubský, R.J.M.Konings, P.Novák, Calculation of Enthalpies of Formation of Actinide Nitrides, J. Nucl. Mater. 344 (2005) 40-44.
- D.Sedmidubský, J.Leitner, Calculation of Thermodynamic Properties of AIII Nitrides, J.Cryst.Growth 286 [1] (2006) 66-70.
- D. Sedmidubský, J. Leitner, O.Beneš, Phase Equilibria Modeling in Bi-Sr-Mn-O System, Calphad 30 [2] (2006) 179-184.
- D.Sedmidubský, J.Leitner, Z.Sofer, Phase Relations in the Ga-Mn-N System, J.Alloy.Compd. 452 (2008) 105-109.
- D.Sedmidubský, J.Leitner, P.Svoboda, Z.Sofer, J.Macháček, Heat Capacity and Phonon Spectra of AIIIN - Experiment and Calculation, J.Therm.Anal.Calorim. 95 (2009) 403-407.
- D.Sedmidubský, R.J.M.Konings, P.Souček, Ab-initio calculations and Phase Diagram Assessments of An-Al Systems (An = U, Np, Pu),J.Nucl.Mater. 397 (2010) 1-7.
- D.Gryaznov, D.Sedmidubský, E. Heifets, Density functional theory calculations on magnetic properties of actinide compounds, J. Phys. Chem. Chem. Phys. 12 (2010) 12273-12278
- D. Sedmidubský, V. Jakeš, O. Jankovský, J. Leitner, Z. Sofer, J. Hejtmánek, Phase Equilibria in Ca-Co-O system, J. Sol. St. Chem. 194 (2012) 199-205
- P.Holba, D.Sedmidubský, Heat capacity equations for nonstoichiometric solids, J. Therm. Anal. Calorim. 113 (2013) 239-245
- D.Sedmidubský, P.Holba, Material properties of nonstoichiometric solids, J. Therm. Anal. Calorim. 120(2015) 183-188